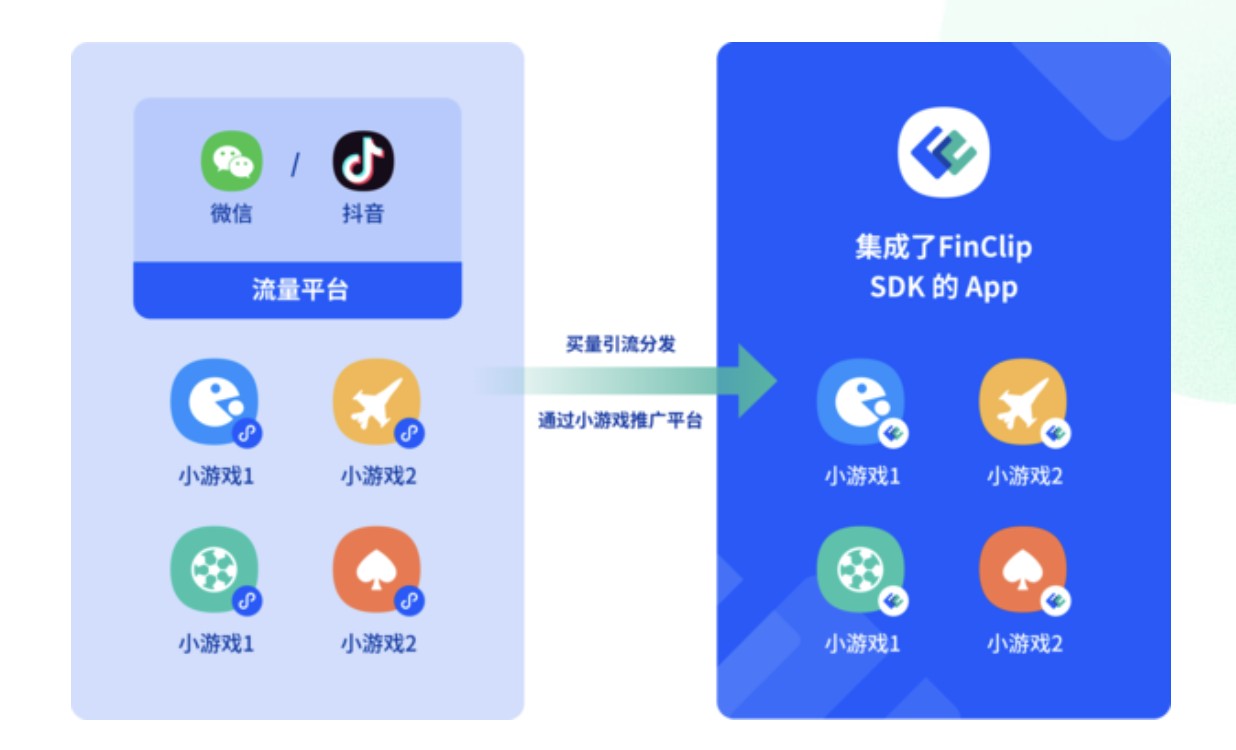
微前端架构如何改变企业的开发模式与效率提升
932
2022-11-25

使用pyGenomeTracks可视化hi-c数据

欢迎关注”生信修炼手册”!
可视化是数据分析中非常重要的一个环节,对于NGS分析数据的可视化,最常用的就是各种基因组浏览器了,既有UCSC, GBrowse等基于web的基因组浏览器,也有igvtools等本地化的图形界面软件。对于Hi-C数据,在前面的文章中也介绍过基于web的WashU Epigenome Browser基因组浏览器和本地化的juicebox软件。
熟练掌握其中一个软件的用法就可以满足大部分的需求了,但是作为一个生信分析的极客,总感觉还是需要一款命令行工具来提高效率。python和R都拥有非常强大的可视化能力,今天介绍一款基于python语言的软件pyGenomeTracks, 一款原汁原味的命令行工具,拥有和基因组浏览器相同的展现形式,网址如下
matrices
采用该软件可视化的效果图如下
和基因组浏览器一样的展现形式,每一层为一个track。该软件采用配置文件的形式来配置需要展示的文件信息,每个需要展示的文件和对应的参数都写在一个标签下,具体写法如下
1. bigwig
2. bedgraph
3. hic
除此之后,还有x-axis和spacer等标签,分别对应x轴和两个tracks之间的空格区域。下方如下
[spacer][x-axis]where = top
编辑好配置文件之后,就可以运行了,用法如下
pyGenomeTracks \--tracks tracks.ini \--region chr2:10,000,000-11,000,000 \--outFileName output.pdf
tracks参数指定配置文件的名称,region参数指定需要可视化的基因组区域,outFileName参数指定输出文件的名称。为了达到美观的效果,有许多的参数需要调整,更多细节请参考官方文档和示例。
一个hi-c数据可视化的效果图如下
通过该软件,可以高效的展示hi-c数据。
·end·
—如果喜欢,快分享给你的朋友们吧—
版权声明:本文内容由网络用户投稿,版权归原作者所有,本站不拥有其著作权,亦不承担相应法律责任。如果您发现本站中有涉嫌抄袭或描述失实的内容,请联系我们jiasou666@gmail.com 处理,核实后本网站将在24小时内删除侵权内容。


发表评论

暂时没有评论,来抢沙发吧~